Slacken paper published in NAR Genomics and Bioinformatics
Our paper on Slacken, a metagenomic classifier based on the Kraken 2 method, was recently published in NAR Genomics and Bioinformatics. In this post I (Johan) would like to break down the main results and what they might mean for Kraken 2 users and microbiologists in a simple way.
The first main result relates to scaling. Kraken 2 is known for storing its library entirely in RAM when running, which makes the memory requirement for the standard library very large (around 100 GB RAM as of RefSeq 224). Very large libraries might need 1 TB of RAM. Slacken decouples library size entirely from the amount of available RAM by dividing the computational tasks into small partitions (thanks to Apache Spark). Instead, it is necessary to have a sufficiently large SSD drive, which is usually a simpler requirement to satisfy. With Slacken, it is possible to classify with the Kraken 2 standard library on a 16-core PC using only 16 GB of RAM. You can do this easily on Linux with Docker by following our tutorial on GitHub.
We are able to scale horizontally as well as down: tests on large clusters (AWS Elastic MapReduce) allowed us to run Slacken on 100s of vCPUs in parallel. This means that we can run the Kraken 2 method with arbitrarily large libraries - quickly - by adding as many commodity machines to a Spark cluster as we need. This removes the upper limit to Kraken 2 library size that was previously imposed by available RAM. Remarkably, for the case with very large libraries and multi-sample classification, the cost per sample of Slacken when running on AWS is comparable to that of Kraken 2.
We are grateful to have received AWS Open Data sponsorship for Slacken, and two metagenomic libraries - the standard one and the very large “rspc” (RefSeq Prefer Complete) one - are currently available to the public. We aim to expand this library collection over time.
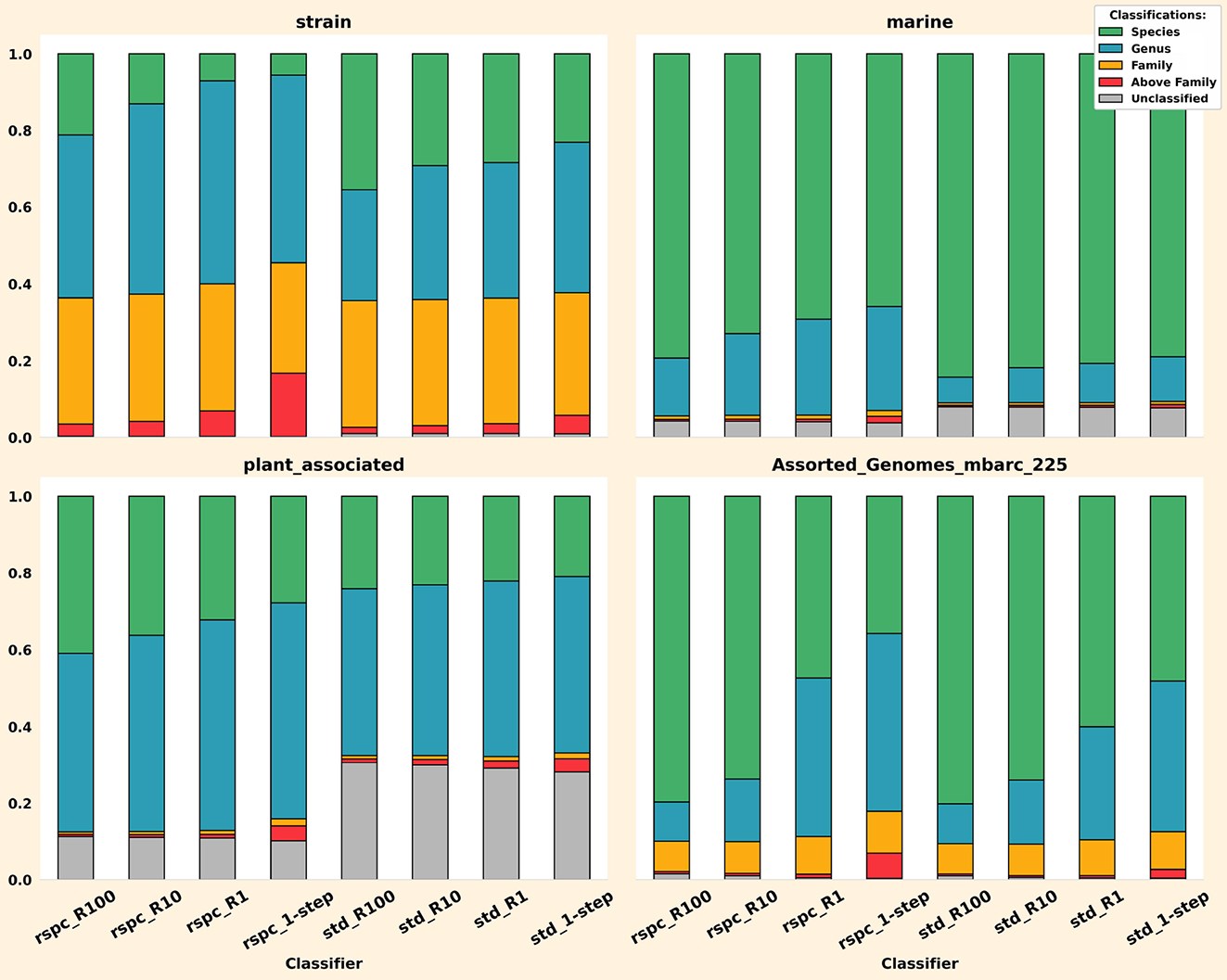
The second main result is custom-built reference libraries for each sample group being classified. We call this sample-tailored minimizer libraries (minimizers being a kind of genomic k-mer that the Kraken 2 method is based on). We exclude, with varying degrees of strictness, genomes that seem to be unrelated to the present sample and instead classify using only the genomes that are apparently present. On average, this greatly increases the amount of species-level read classifications we get (shown in the figure above, e.g. in the progression from rspc_R1 to rspc_R100), which means classifications are more specific and thus more informative. It also improves Bracken outputs (yes, Slacken is compatible with Bracken and implements the functions of bracken-build).
In the paper we study sample-tailored minimizer libraries, which we also call 2-step classification, on the CAMI2 as well as synthetic samples. By using the tutorial, you can try it yourself on your local PC (or in the cloud on AWS EMR, using a different tutorial).
For questions, comments or feedback, all of which we would very much appreciate, please get in touch at: johan@jnpsolutions.io.